
1.2 多孔阳极氧化膜形成机理简要回顾
1.2.1 场致助溶理论
场致助溶(FAD)理论认为阳极氧化过程是在电解液/氧化物(E/O)和氧化物/金属(O/M)这两个界面上产生的[3,4]。阳极氧化分为三个步骤。
(1)图1.1是多孔氧化膜在恒流条件下形成过程记录的阳极氧化曲线示意图。在阳极氧化过程中,在电场E的作用下阳极金属基片表面首先生成一层致密的氧化膜(即图1.1的Ⅰ阶段),致密膜的厚度随着Ti4+和O2-由于高电场的作用不断迁移而不断增厚的。原因就是离子迁移使两界面处发生Ti4+ + 2O2-→TiO2。图1.2所示为FAD理论提出的纳米管的形成过程,新的氧化物按照图1.2a示意图方式不断生长,图1.2b为FAD的示意图。
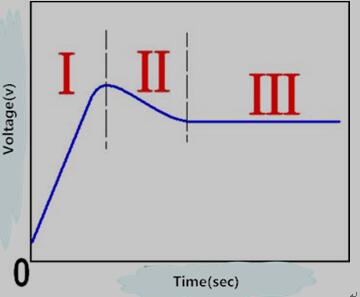
图1.1 恒流条件下多孔氧化膜阳极氧化曲线示意图
(2)众所周知,钛的阳极氧化机制类似于铝,FAD理论就是类比多孔阳极氧化铝膜形成理论提出的。多孔阳极氧化铝膜上发生Al2O3 + 6H + → 2Al3 + + 3H2O,导致氧化膜形成孔洞,猜想阳极氧化钛薄膜上也发生类似溶解反应TiO2 + 6F− + 4H + →[TiF6]2− + 2H2O,这是TNTAs形成的起点。然而,这一猜想并未在实验中获得验证[5]。但猜想两个界面上的氧化物由于氟离子的作用,氧化物中的Ti被夺走以一种结合更稳定的形式——[TiF6]2-进入溶液中,氧阴离子不能单独形成氧化物,所以界面上就发生了从上至下的溶解挖洞(如图1.2b所示),从此开始了孔道形成之路。注意在这时,氧化物的生长仍在继续。
图1.2 a)阳极氧化物生成示意图;b)场致助溶示意图
(3)当氧化物溶解和氧化物生成反应达到平衡时, 孔道底部的阻挡层厚度文持不变,如图1.1中的第Ⅲ阶段所示。此时阻挡层厚度d不变,外加场强E不变,由E=U/d所得,电压U不变即恒电压。
然而,张少瑜等[6]的团队的报道称,三层TNTAs形成过程相应的阳极氧化曲线并不能被FAD理论所验证。因为溶解方程(TiO2 + 6F-+ 4H+ → [TiF6]2− + 2H2O)仅仅发生在电解液/氧化物(E/O)界面上,并不能提供电荷在阻挡层中建立电路。换句话说,穿过金属基体的FAD电流是不可能存在的,可溶产物[TiF6]2−阴离子仅仅进入电解液中而不能在阻挡层中流动,FAD反应本质上是化学的,不能产生阳极氧化电流。此外还应注意到,溶解方程是酸催化,然而大多数氢离子在电场驱动下在阴极附近聚集,然后以氢气的形式释放出去,结果导致在孔底部没有足够的氢离子参与溶解方程,这也验证了用来在阻挡层挖洞的FAD电流是不存在的。我们认为虽然整个阳极氧化过程在电解液/氧化物(E/O)界面上都会发生溶解反应,但是阳极氧化电流的突然变化是很难通过溶解反应来解释的。FAD理论还有很多不能解释的地方,比如元胞之间缝隙的生成,但是它对孔洞加深的解释还是为探索TNTAs形成机理的研究提供了一种思路。
1.2.2 粘性流动模型
2006年,曼彻斯特大学的科学家Skeldon等[7]提出了一个新颖的粘性流动模型。他们使用钨示踪,将钨掺入到溅射沉积的铝衬底上,以便研究阳极氧化铝在磷酸、丙二酸和草酸中孔隙的形成。图1.3是在温度为293K下,在0.3M丙二酸电解液中恒流条件下(电流密度为5mA•cm-2)阳极氧化50nm厚的Al的扫描电镜显微图,目的是检测钨示踪剂的位置。图1.3a是阳极氧化前铝基体的扫描电镜,图1.3b是阳极氧化了250s铝基体的扫面电镜,图1.3c是阳极氧化了400s铝基体的扫描电镜。从图1.3a到图1.3b和图1.3c的变化中可以发现,阻挡区域内的钨层示踪剂停滞在孔下方与相邻的孔壁区域中。并且在电解液中并没有检测到损失的钨。此外,在非富含电解质的电解液中后续阳极氧化过程中,还发现了18O离子被保留在薄膜内。这些发现表明在阳极氧化过程中,氧化膜的溶解可近似忽略,这似乎违背了FAD理论,因为FAD理论认为氧化物溶解对多孔阳极薄膜生长过程具有极大的影响。 在PEG溶液中TiO2纳米管的形成机理研究(2):http://www.youerw.com/huaxue/lunwen_21309.html