- 上一篇:HPLC法测定水环境中微量PPCPs研究
- 下一篇:农药残留测试条的批量生产工艺研究
1.2.1.1 均相沉淀成核机理
该机理是由Fitch和Tsai等人于1969年第一次提出的[18,20]。Goodwin等人后来又做了部分补充。他们认为,聚合反应发生在水相中,引发剂分解后生成初级自由基,并和强亲水性单体(例如MMA)结合,生成大量的齐聚物自由基,链增长较快,随着齐聚物自由基的聚合度不断增大,当体系中的自由基浓度达到一定值时,活性链发生自身弯曲缠结,不溶于水相,从而析出形成带电荷的初始胶乳粒子,由于这些粒子的比表面积比较大且电荷密度较低,粒子之间的静电排斥力较弱,以致粒子变得不稳定,从而开始聚结,初始乳胶粒子慢慢相互聚集最终生成稳定的胶粒。同时,微球粒子被单体溶胀,最终粒子粒径不断变大[21]。均相成核机理的具体过程如图1.1(a)。
1.2.1.2 齐聚物胶束成核机理
Goodwall等人以KPS为引发剂,通过透射电镜(TEM)、SEM、凝胶渗透色谱(GPC)等方法研究了苯乙烯的无皂乳液聚合,并于1977年提出了齐聚物胶束成核机理。该理论认为:反应初期时引发速度比链增长速度要快,水相中产生大量的齐聚物链,链端处带着有一定活性的亲水性基团,当这些短链聚合物不断地增长达到一定的聚合度和浓度时,将产生具有一定表面活性的大分子自由基,在水相中形成低聚物胶束粒子;当单体或是链增长自由基慢慢扩散到了胶束中,这些胶束粒子变得不再稳定,颗粒粒径开始变大,稳定性也会变差,颗粒之间发生聚集后生成稳定的乳胶粒子[22]。其成核机理如图1.2(b)。
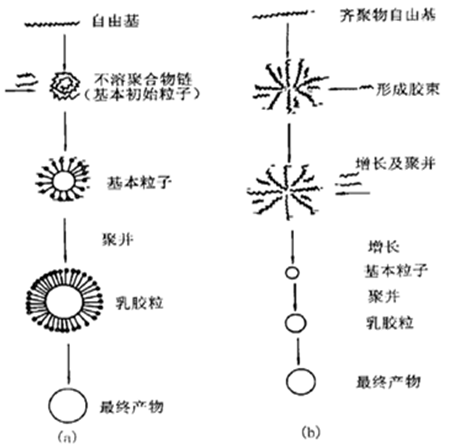
图1.1 成核机理示意图
(a)均相沉淀成核;(b)齐聚物胶束成核
1.2.2 无皂乳液聚合方法
无皂乳液聚合方法大致有以下几种:(1)引发剂碎片法[23](2)水溶性单体共聚法(3)反应性乳化剂共聚法[24](4)超声无皂乳液聚合法。
1.2.2.1 引发剂碎片法
用引发剂碎片法通过采用不同的引发剂制得含有多种功能基团的乳胶粒子,从而控制乳胶颗粒表面的性质,制得多种高分子功能聚合物微球。这种方法中,经引发剂分解得到离子基团,体系中的乳胶粒子只能依靠离子基团间的静电作用来稳定,聚合反应速率缓慢,最终聚合物乳液固含量偏低,往往不超过10%[25],得到的乳胶粒子表面电荷数目较少,稳定性一般。
1.2.2.2 水溶性单体共聚法
较为常见的水溶性共聚单体有羧酸类、酰胺类、磺酸类单体等。引入水溶性共聚单体有两大优点,一是可以增加反应体系中水相中的单体浓度,加快聚合反应速率;二是代替乳化剂起到稳定乳液的作用,使乳液更稳定。
1.2.2.3 反应性乳化剂共聚法
选用反应性乳化剂可克服传统乳化剂总是容易迁移并残留在产物中影响多种应用性能的缺陷,也可增加乳液的固含量,是一种十分有效的聚合方法。
1.2.2.4 超声无皂乳液聚合法
利用超声波可以替代传统无皂乳液聚合中的引发剂,更好更快地制备出所需要的无皂乳液。其原理是超声波作用于液体介质时会使无皂乳液发生“空化”,局部温度达5000K之高,这些能量可促进反应,提高聚合速率。
1.2.3 无皂乳液聚合的稳定性
无皂乳液聚合与传统乳液聚合相比,体系中缺少了乳化剂的稳定作用,乳胶粒子主要的稳定方法是依靠结合在高分子链或是高分子链的端基上的离子基团或亲水性基团等。提高无皂乳液聚聚合体系稳定性的方法有:采用齐聚物分散体系、提高微球粒子表面的亲水性[26]和添加有机溶剂[27]、添加无机粉末[28]以及选择适当的制备工艺[29]等。
-
WnCu团簇的结构和性能研究
-
ZnO/碳纳米复合纤维的制备...
-
sol-gel制备Sr掺杂的LaFeO3纳米颗粒和气敏性能
-
碳化硅改性水性聚氨酯的研究
-
栗壳吸附质生物质活性炭的制备与应用
-
WO3-PbO2/C混合电容器的组装及性能研究
-
Cu配合物的合成与分离乙烯/乙烷的性能研究
上市公司股权结构对经营绩效的影响研究
巴金《激流三部曲》高觉新的悲剧命运
中国传统元素在游戏角色...
g-C3N4光催化剂的制备和光催化性能研究
现代简约美式风格在室内家装中的运用
江苏省某高中学生体质现状的调查研究
NFC协议物理层的软件实现+文献综述
高警觉工作人群的元情绪...
C++最短路径算法研究和程序设计
浅析中国古代宗法制度